


 Sign Out
Sign Out

Pharmacology: Pharmacodynamics: Mechanism of action: Fluticasone furoate and vilanterol represent two classes of medications (a synthetic corticosteroid and a selective, long-acting beta2-receptor agonist).
Pharmacodynamic effects: Fluticasone furoate: Fluticasone furoate is a synthetic trifluorinated corticosteroid with potent anti-inflammatory activity. The precise mechanism through which fluticasone furoate affects asthma and COPD symptoms is not known. Corticosteroids have been shown to have a wide range of actions on multiple cell types (e.g. eosinophils, macrophages, lymphocytes) and mediators (e.g. cytokines and chemokines involved in inflammation).
Vilanterol trifenatate: Vilanterol trifenatate is a selective long-acting, beta2-adrenergic agonist (LABA).
The pharmacologic effects of beta2-adrenoceptor agonist drugs, including vilanterol trifenatate, are at least in part attributable to stimulation of intracellular adenylate cyclase, the enzyme that catalyzes the conversion of adenosine triphosphate (ATP) to cyclic-3',5'-adenosine monophosphate (cyclic AMP). Increased cyclic AMP levels cause relaxation of bronchial smooth muscle and inhibition of release of mediators of immediate hypersensitivity from cells, especially from mast cells.
Molecular interactions occur between corticosteroids and LABAs, whereby steroids activate the beta2-receptor gene, increasing receptor number and sensitivity and LABAs prime the glucocorticoid receptor for steroid-dependent activation and enhance cell nuclear translocation. These synergistic interactions are reflected in enhanced anti-inflammatory activity, which has been demonstrated in vitro and in vivo in a range of inflammatory cells relevant to the pathophysiology of both asthma and COPD. In peripheral blood mononuclear cells from subjects with COPD, a larger anti-inflammatory effect was seen in the presence of the combination of fluticasone furoate/vilanterol compared with fluticasone furoate alone at concentrations achieved with clinical doses.
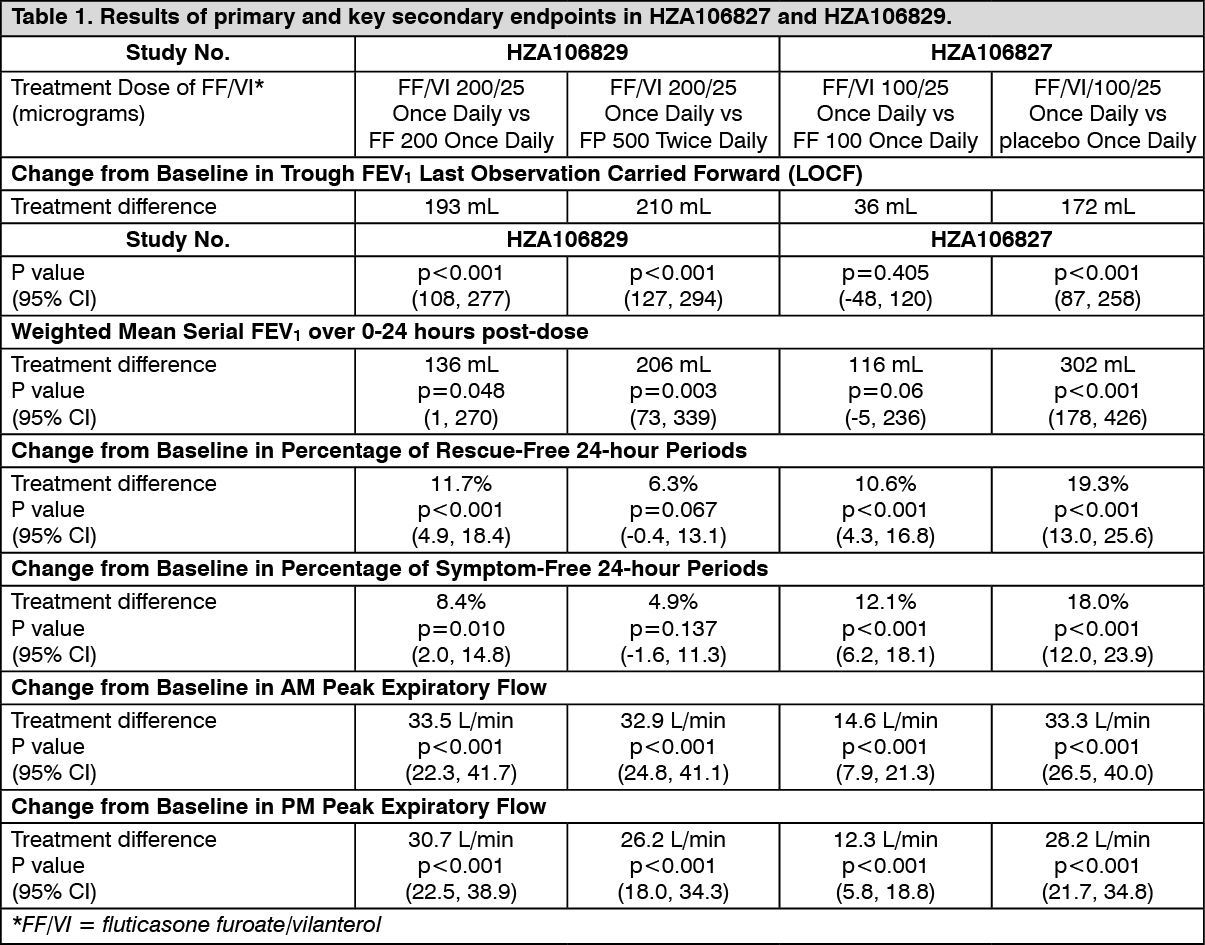
Clinical efficacy and safety:Asthma: Three phase III randomised, double-blind studies (HZA106827, HZA106829 and HZA106837) of different durations evaluated the safety and efficacy of fluticasone furoate/vilanterol in adult and adolescent patients with persistent asthma. All subjects were using an ICS (Inhaled corticosteroid) with or without LABA for at least 12 weeks prior to visit 1. In HZA106837 all patients had at least one exacerbation that required treatment with oral corticosteroids in the year prior to visit 1. HZA106827 was 12 weeks in duration and evaluated the efficacy of fluticasone furoate/vilanterol 100/25 micrograms [n=201] and FF 100 micrograms [n=205]) compared with placebo [n=203], all administered once daily. HZA106829 was 24 weeks in duration and evaluated the efficacy of fluticasone furoate/vilanterol 200/25 micrograms [n=197] and FF 200 micrograms [n=194]) both administered once daily compared with FP 500 micrograms twice daily [n=195].
In HZA106827/HZA106829 the co-primary efficacy endpoints were changed from baseline in clinic visit trough (pre-bronchodilator and pre-dose) FEV1 at the end of the treatment period in all subjects and weighted mean serial FEV1 over 0-24 hours post-dose calculated in a subset of subjects at the end of the treatment period. Change from baseline in the percentage of rescue-free 24-hour periods during treatment was a powered secondary endpoint. Results for the primary and key secondary endpoints in these studies are described in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageHZA106837 was of variable treatment duration (from a minimum of 24 weeks to a maximum of 76 weeks with the majority of patients treated for at least 52 weeks). In HZA106837 patients were randomised to receive either fluticasone furoate/vilanterol 100/25 micrograms [n=1009] or FF 100 micrograms [n=1010] both administered once daily. In HZA106837 the primary endpoint was the time to first severe asthma exacerbation. A severe asthma exacerbation was defined as deterioration of asthma requiring the use of systemic corticosteroids for at least 3 days or an inpatient hospitalization or emergency department visit due to asthma that required systemic corticosteroids. Adjusted mean change from baseline in trough FEV1 was also evaluated as a secondary endpoint.
In HZA106837 the risk of experiencing a severe asthma exacerbation in patients receiving fluticasone furoate/vilanterol 100/25 micrograms was reduced by 20% compared with FF 100 micrograms alone (hazard ratio 0.795, p=0.036 95% CI 0.642, 0.985). The rate of severe asthma exacerbations per patient per year was 0.19 in the FF 100 micrograms group (approximately 1 in every 5 years) and 0.14 in the fluticasone furoate/vilanterol 100/25 micrograms group (approximately 1 in every 7 years). The ratio of the exacerbation rate for fluticasone furoate/vilanterol 100/25 micrograms versus FF 100 micrograms was 0.755 (95% CI 0.603, 0.945). This represents a 25% reduction in the rate of severe asthma exacerbations for subjects treated with fluticasone furoate/vilanterol 100/25 micrograms compared with FF 100 micrograms(p=0.014). The 24-hour bronchodilator effect of fluticasone furoate/vilanterol was maintained throughout a one-year treatment period with no evidence of loss in efficacy (no tachyphylaxis). Fluticasone furoate/vilanterol 100/25 micrograms consistently demonstrated 83 mL to 95 mL improvements in trough FEV1 at weeks 12, 36 and 52 and Endpoint compared with FF 100 micrograms (p<0.001 95% CI 52, 126 mL at Endpoint). Forty four percent of patients in the fluticasone furoate/vilanterol 100/25 micrograms group were well controlled (ACQ7 ≤0.75) at end of treatment compared to 36% of subjects in the FF 100 micrograms group (p<0.001 95% CI 1.23, 1.82).
Studies versus salmeterol/fluticasone propionate combinations: In a 24-week study (HZA113091) in adult and adolescent patients with uncontrolled persistent asthma both fluticasone furoate/vilanterol 100/25 micrograms given once daily in the evening and salmeterol/FP 50/250 micrograms given twice daily demonstrated improvements from baseline in lung function. Adjusted mean treatment increases from baseline in weighted mean 0-24 hours FEV1 of 341 mL (fluticasone furoate/vilanterol) and 377 mL (salmeterol/FP) demonstrated an overall improvement in lung function over 24 hours for both treatments. The adjusted mean treatment difference of 37 mL between the groups was not statistically significant (p=0.162). For trough FEV1 subjects in the fluticasone furoate/vilanterol group achieved a LS mean change from baseline of 281 mL and subjects in salmeterol/FP group a change of 300 mL; (the difference in adjusted mean of 19 mL (95% CI: -0.073, 0.034) was not statistically significant (p=0.485).
A randomised, double-blind, parallel-group, 24-week study (201378) was conducted to demonstrate non-inferiority (using a margin of -100 mL for trough FEV1) of fluticasone furoate/vilanterol 92/22 once daily to salmeterol/FP 50/250 twice daily in adults whose asthma was well controlled following 4 weeks of treatment with open-label salmeterol/FP 50/250 twice daily (N=1504). Subjects randomised to once-daily FF/VI maintained lung function comparable with those randomised to twice-daily salmeterol/FP [difference in trough FEV1 of +19 mL (95% CI: -11, 49)].
No comparative studies versus salmeterol/FP or versus other ICS/LABA combinations have been conducted to appropriately compare the effects of asthma exacerbations.
Fluticasone furoate monotherapy: A 24-week randomised, double-blind placebo-controlled study (FFA112059) evaluated the safety and efficacy of FF 100 micrograms once daily [n=114] and FP 250 micrograms twice daily [n=114] versus placebo [n=115] in adult and adolescent patients with persistent asthma. All subjects had to have been on a stable dose of an ICS for at least 4 weeks prior to visit 1 (screening visit) and the use of LABAs was not permitted within 4 weeks of visit 1. The primary efficacy endpoint was change from baseline in clinic visit trough (pre-bronchodilator and pre-dose) FEV1 at the end of the treatment period. Change from baseline in the percentage of rescue-free 24-hour periods during the 24-week treatment period was a powered secondary. At the 24-week time point FF and FP increased trough FEV1 by 146 mL (95% CI 36, 257 mL, p=0.009) and 145 mL (95% CI 33, 257 mL, p=0.011) respectively compared to placebo. FF and FP both increased the percentage of 24-hour rescue-free periods by 14.8% (95% CI 6.9, 22.7, p<0.001) and 17.9% (95% CI 10.0, 25.7, p<0.001) respectively versus placebo.
Allergen challenge study: The bronchoprotective effect of fluticasone furoate/vilanterol 100/25 micrograms on the early and late asthmatic response to inhaled allergen was evaluated in a repeat dose, placebo-controlled four-way crossover study (HZA113126) in patients with mild asthma. Patients were randomized to receive fluticasone furoate/vilanterol 100/25 micrograms, FF 100 micrograms, vilanterol 25 micrograms or placebo once daily for 21 days followed by challenge with allergen 1 hour after the final dose. The allergen was house dust mite, cat dander, or birch pollen; the selection was based on individual screening tests. Serial FEV1 measurements were compared with pre-allergen challenge values taken after saline inhalation (baseline). Overall, the greatest effects on the early asthmatic response were seen with fluticasone furoate/vilanterol 100/25 micrograms compared with FF 100 micrograms or vilanterol 25 micrograms alone. Both fluticasone furoate/vilanterol 100/25 micrograms and FF 100 micrograms virtually abolished the late asthmatic response compared with vilanterol alone. Fluticasone furoate/vilanterol 100/25 micrograms provided significantly greater protection against allergen-induced bronchial hyper-reactivity compared with monotherapies FF and vilanterol as assessed on Day 22 by methacholine challenge.
100/25 mcg: Chronic Obstructive Pulmonary Disease: The COPD clinical development programme included a 12-week (HZC113107), two 6-month (HZC112206, HZC112207), two one-year (HZC102970, HZC102871) and one long term study (SUMMIT). These were randomised controlled studies in patients with a clinical diagnosis of COPD. These studies included measures of lung function, dyspnoea and moderate and severe exacerbations.
Six-month studies: HZC112206 and HZC112207 were 24-week randomised, double-blind, placebo-controlled, parallel-group studies comparing the effect of the combination to vilanterol and FF alone and placebo. HZC112206 evaluated the efficacy of fluticasone furoate/vilanterol 50/25 micrograms [n=206] and fluticasone furoate/vilanterol 100/25 micrograms [n=206] compared with FF 100 micrograms [n=206], vilanterol 25 micrograms [n=205] and placebo [n=207], all administered once daily.
HZC112207 evaluated the efficacy of fluticasone furoate/vilanterol 100/25 micrograms [n=204] and fluticasone furoate/vilanterol 200/25 micrograms [n=205] compared with FF 100 micrograms [n=204], FF 200 micrograms [n=203] and vilanterol 25 micrograms [n=203] and placebo [n=205], all administered once daily.
All patients were required to have a smoking history of at least 10 pack years; a post-salbutamol FEV1/FVC ratio less than or equal to 0.70; post-salbutamol FEV1 less than or equal to 70% predicted and have a Modified Medical Research Council (mMRC) dyspnea score ≥2 (scale 0-4) at screening. At screening, the mean pre-bronchodilator FEV1 was 42.6% and 43.6% predicted, and the mean reversibility was 15.9% and 12.0% in HZC112206 and HZC112207, respectively. The co-primary endpoints in both studies were weighted mean FEV1 from zero to 4 hours post-dose at Day 168 and change from baseline in pre-dose trough FEV1 at Day 169.
In an integrated analysis of both studies, fluticasone furoate/vilanterol 100/25 micrograms showed clinically meaningful improvements in lung function. At Day 169 fluticasone furoate/vilanterol 100/25 micrograms and vilanterol increased adjusted mean trough FEV1 by 129 mL (95% CI: 91, 167 mL, p<0.001) and 83 mL (95% CI: 46, 121 mL, p<0.001) respectively compared to placebo. Fluticasone furoate/vilanterol 100/25 micrograms increased trough FEV1 by 46 mL compared to vilanterol (95% CI: 8, 83 mL, p= 0.017). At Day 168 fluticasone furoate/vilanterol 100/25 micrograms and vilanterol increased adjusted mean weighted mean FEV1 over 0-4 hours by 193 mL (95% CI: 156, 230 mL, p<0.001) and 145 mL (95% CI: 108, 181 mL, p<0.001) respectively compared to placebo. Fluticasone furoate/vilanterol 100/25 micrograms increased adjusted mean weighted mean FEV1 over 0-4 hours by 148 mL compared to FF alone (95% CI: 112, 184 mL, p<0.001).
12-month studies: Studies: HZC102970 and HZC102871 were 52-week randomised, double-blind, parallel-group, studies comparing the effect of fluticasone furoate/vilanterol 200/25 micrograms, fluticasone furoate/vilanterol 100/25 micrograms, fluticasone furoate/vilanterol 50/25 micrograms with vilanterol 25 micrograms, all administered once daily, on the annual rate of moderate/severe exacerbations in subjects with COPD with a smoking history of at least 10 pack years and a post-salbutamol FEV1/FVC ratio less than or equal to 0.70 and post-salbutamol FEV1 less than or equal to 70% predicted and documented history of ≥1 COPD exacerbation that required antibiotics and/or oral corticosteroids or hospitalisation in the 12 months prior to visit 1. The primary endpoint was the annual rate of moderate and severe exacerbations. Moderate/severe exacerbations were defined as worsening symptoms that required treatment with oral corticosteroids and/or antibiotics or in-patient hospitalisation. Both studies had a 4-week run-in period during which all subjects received open-label salmeterol/FP 50/250 twice daily to standardise COPD pharmacotherapy and stabilise disease prior to randomisation to blinded study medication for 52 weeks. Prior to run-in, subjects discontinued use of previous COPD medications except short-acting bronchodilators. The use of concurrent inhaled long-acting bronchodilators (beta2-agonist and anticholinergic), ipratropium/salbutamol combination products, oral beta2-agonists, and theophylline preparations were not allowed during the treatment period. Oral corticosteroids and antibiotics were allowed for the acute treatment of COPD exacerbations with specific guidelines for use. Subjects used salbutamol on an as-needed basis throughout the studies.
The results of both studies showed that treatment with fluticasone furoate/vilanterol 100/25 micrograms once daily resulted in a lower annual rate of moderate/severe COPD exacerbations compared with vilanterol (Table 2). Reductions in risk of time to first moderate or severe exacerbation and rate of exacerbations requiring corticosteroid use were also observed with fluticasone furoate/vilanterol 100/25 micrograms once daily compared with vilanterol. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn an integrated analysis of HZC102970 and HZC102871 at week 52, an improvement was seen when comparing the fluticasone furoate/vilanterol 100/25 micrograms versus vilanterol 25 micrograms in adjusted mean trough FEV1 (42 mL 95% CI: 19, 64 mL, p<0.001). The 24-hour bronchodilator effect of fluticasone furoate/vilanterol was maintained from the first dose throughout a one-year treatment period with no evidence of loss in efficacy (no tachyphylaxis).
Overall, across the two studies combined 2009 (62%) patients had cardiovascular history/risk factors at screening. The incidence of cardiovascular history/risk factors was similar across the treatment groups with patients most commonly suffering from hypertension (46%), followed by hypercholesterolemia (29%) and diabetes mellitus (12%). Similar effects in reduction of moderate and severe exacerbations were observed in this subgroup as compared with the overall population. In patients with a cardiovascular history/risk factors, fluticasone furoate/vilanterol 100/25 micrograms resulted in a significantly lower annual rate of moderate/severe COPD exacerbations compared with vilanterol (adjusted mean annual rates of 0.83 and 1.18 respectively, 30% reduction (95% CI: 16, 42%, p<0.001). Improvements were also seen in this subgroup at week 52 when comparing the fluticasone furoate/vilanterol 100/25 micrograms vs. vilanterol 25 micrograms in adjusted mean trough FEV1 (44 mL 95% CI: 15, 73 mL, (p=0.003)).
Long-term study: SUMMIT was a multi-centre, randomised, double-blind study evaluating the effect on survival of fluticasone furoate/vilanterol 100/25 micrograms compared with placebo in 16,568 subjects. Subjects were treated for up to 4 years (mean 1.7 years) with either fluticasone furoate/vilanterol 100/25 micrograms, fluticasone furoate 100 micrograms, vilanterol 25 micrograms, or placebo. All subjects had COPD with moderate airflow limitation (≥50% and ≤70% predicted FEV1) and a history of, or an increased risk of, cardiovascular disease. Survival with fluticasone furoate/vilanterol was not significantly improved compared with placebo (HR 0.878; 95% CI: 0.739, 1.042; p=0.137), fluticasone furoate (HR 0.964; 95% CI: 0.808, 1.149; p=0.681) or vilanterol (HR 0.912; 95% CI: 0.767, 1.085; p=0.299). All-cause mortality was: fluticasone furoate/vilanterol, 6.0%; placebo, 6.7%; fluticasone furoate, 6.1%; vilanterol, 6.4%).
Fluticasone furoate/vilanterol slowed the rate of decline in lung function as measured by FEV1, by 8 mL/year compared with placebo (95% CI: 1, 15; p=0.019). There was no impact (0 mL/year; 95% CI: -6, 7; p=0.913) on the rate of decline for fluticasone furoate/vilanterol compared with fluticasone furoate; there was a difference of 10 mL/year for fluticasone furoate/vilanterol compared with vilanterol (95% CI: 3, 16; p=0.004). The mean rate of decline in FEV1 was: fluticasone furoate/vilanterol, 38 mL/year; placebo, 46 mL/year; fluticasone furoate, 38 mL/year; vilanterol, 47 mL/year.
The risk of a cardiovascular composite event (on-treatment cardiovascular death, myocardial infarction, stroke, unstable angina, or transient ischemic attack) with fluticasone furoate/vilanterol was not significantly lower than placebo (HR 0.926; 95% CI: 0.750, 1.143; p=0.475), fluticasone furoate (HR 1.033; 95% CI: 0.834, 1.281; p=0.763) or vilanterol (HR 0.938; 95% CI: 0.761, 1.155; p=0.545). The incidence of cardiovascular composite events was: fluticasone furoate/vilanterol, 4.2%; placebo, 4.2%; fluticasone furoate, 3.9%; vilanterol 4.4%.
Fluticasone furoate/vilanterol demonstrated a larger mean change from baseline in post-bronchodilator FEV1 at Day 360 compared with placebo (89 mL; 95% CI: 76, 102; p<0.001), fluticasone furoate (40 mL; 95% CI: 27, 53; p<0.001), and vilanterol (26 mL; 95% CI: 13, 39; p<0.001). The adjusted mean change from baseline was: fluticasone furoate/vilanterol 50 mL, placebo, -39 mL; fluticasone furoate, 9 mL; vilanterol, 24 mL.
Fluticasone furoate/vilanterol reduced the annual rate of moderate or severe exacerbations by 29% (95% CI: 22, 35; p<0.001) compared with placebo, by 19% compared with fluticasone furoate (95% CI: 12, 26; p<0.001) and by 21% compared with vilanterol (95% CI: 14, 28; p<0.001). The annual rate of moderate or severe exacerbations was 0.25 for fluticasone furoate/vilanterol, 0.35 for placebo, 0.31 for fluticasone furoate, and 0.31 for vilanterol.
Fluticasone furoate/vilanterol reduced the annual rate of severe exacerbations (i.e. requiring hospitalisation) by 27% (95% CI: 13, 39; p<0.001) compared with placebo, by 11% compared with fluticasone furoate (95% CI: -6, 25; p=0.204) and by 9% compared with vilanterol (95% CI: -8, 24; p=0.282). The annual rate of exacerbations requiring hospitalisation was 0.05 for fluticasone furoate/vilanterol, 0.07 for placebo, 0.06 for fluticasone furoate, and 0.06 for vilanterol.
Studies versus salmeterol/fluticasone propionate combinations: In a 12-week study (HZC113107) in COPD patients both fluticasone furoate/vilanterol 100/25 micrograms given once daily in the morning and salmeterol/FP 50/500 micrograms given twice daily, demonstrated improvements from baseline in lung function. Adjusted mean treatment increases from baseline in weighted mean 0-24 hours FEV1 of 130 mL (fluticasone furoate/vilanterol) and 108 mL (salmeterol/FP) demonstrated an overall improvement in lung function over 24 hours for both treatments. The adjusted mean treatment difference of 22 mL (95% CI: -18, 63 mL) between the groups was not statistically significant (p=0.282). The adjusted mean change from baseline in trough FEV1 on Day 85 was 111 mL in the fluticasone furoate/vilanterol group and 88 mL in the salmeterol/FP group; the 23 mL (95% CI: -20, 66) difference between the treatment groups was not clinically meaningful or statistically significant (p=0.294). No comparative studies versus salmeterol/FP or versus other established bronchodilators have been conducted to appropriately compare the effects on COPD exacerbations.
Paediatric population: The European Medicines Agency has waived the obligation to submit the results of studies with Relvar Ellipta in all subsets of the paediatric population in COPD (see Dosage & Administration for information on paediatric use).
The European Medicines Agency has deferred the obligation to submit the results of studies with Relvar Ellipta in one or more subsets of the paediatric population in asthma (see Dosage & Administration for information on paediatric use).
Pharmacokinetics: Absorption: The absolute bioavailability for fluticasone furoate and vilanterol when administered by inhalation as fluticasone furoate/vilanterol was on average 15.2% and 27.3%, respectively. The oral bioavailability of both fluticasone furoate and vilanterol was low, on average 1.26% and <2%, respectively. Given this low oral bioavailability, systemic exposure for fluticasone furoate and vilanterol following inhaled administration is primarily due to absorption of the inhaled portion of the dose delivered to the lung.
Distribution: Following intravenous dosing, both fluticasone furoate and vilanterol are extensively distributed with average volumes of distribution at steady state of 661 L and 165 L, respectively.
Both fluticasone furoate and vilanterol have a low association with red blood cells. In vitro plasma protein binding in human plasma of fluticasone furoate and vilanterol was high, on average >99.6% and 93.9%, respectively. There was no decrease in the extent of in vitro plasma protein binding in subjects with renal or hepatic impairment.
Fluticasone furoate and vilanterol are substrates for P-glycoprotein (P-gp), however, concomitant administration of fluticasone furoate/vilanterol with P-gp inhibitors is considered unlikely to alter fluticasone furoate or vilanterol systemic exposure since they are both well absorbed molecules.
Biotransformation: Based on in vitro data, the major routes of metabolism of both fluticasone furoate and vilanterol in human are mediated primarily by CYP3A4.
Fluticasone furoate is primarily metabolised through hydrolysis of the S-fluoromethyl carbothioate group to metabolites with significantly reduced corticosteroid activity. Vilanterol is primarily metabolised by O-dealkylation to a range of metabolites with significantly reduced β1- and β2-agonist activity.
Elimination: Following oral administration, fluticasone furoate was eliminated in humans mainly by metabolism with metabolites being excreted almost exclusively in faeces, with <1% of the recovered radioactive dose eliminated in the urine.
Following oral administration, vilanterol was eliminated mainly by metabolism followed by excretion of metabolites in urine and faeces approximately 70% and 30% of the radioactive dose respectively in a human radiolabel study conducted by the oral route. The apparent plasma elimination half-life of vilanterol following single inhaled administration of fluticasone furoate/vilanterol was, on average, 2.5 hours. The effective half-life for accumulation of vilanterol, as determined from inhalation administration of repeat doses of vilanterol 25 micrograms, is 16.0 hours in subjects with asthma and 21.3 hours in subjects with COPD.
Paediatric population: In adolescents (12 years or older), there are no recommended dose modifications.
The pharmacokinetics of fluticasone furoate/vilanterol in patients less than 12 years of age has not been studied. The safety and efficacy of fluticasone furoate/vilanterol in children under the age of 12 years has not yet been established.
Special populations: Elderly patients (>65 years old): The effects of age on the pharmacokinetics of fluticasone furoate and vilanterol were determined in phase III studies in COPD and asthma. There was no evidence for age (12-84) to affect the pharmacokinetics of fluticasone furoate and vilanterol in subjects with asthma.
In subjects with asthma and subjects with COPD there are no recommended dose modifications.
100/25 mcg: There was no evidence for age to affect the pharmacokinetics of fluticasone furoate in subjects with COPD while there was an increase (37%) in AUC of vilanterol over the observed age range of 41 to 84 years. For an elderly subject (aged 84 years) with low body weight (35 kg) vilanterol AUC(0-24) is predicted to be 35% higher than the population estimate (subject with COPD aged 60 years and body weight of 70 kg), whilst Cmax was unchanged. These differences are unlikely to be of clinical relevance.
Renal impairment: A clinical pharmacology study of fluticasone furoate/vilanterol showed that severe renal impairment (creatinine clearance <30 mL/min) did not result in significantly greater exposure to fluticasone furoate or vilanterol or more marked corticosteroid or beta2-agonist systemic effects compared with healthy subjects.
No dose adjustment is required for patients with renal impairment.
The effects of haemodialysis have not been studied.
Hepatic impairment: Following repeat dosing of fluticasone furoate/vilanterol for 7 days, there was an increase in fluticasone furoate systemic exposure (up to three-fold as measured by AUC(0–24)) in subjects with hepatic impairment (Child-Pugh A, B or C) compared with healthy subjects. The increase in fluticasone furoate systemic exposure in subjects with moderate hepatic impairment (Child-Pugh B; fluticasone furoate/vilanterol 200/25 micrograms) was associated with an average 34% reduction in serum cortisol compared with healthy subjects. In subjects with severe hepatic impairment (Child Pugh C) that received a lower dose of 100/2.5 micrograms there was no reduction in serum cortisol. For patients with moderate or severe hepatic impairment the maximum dose of 100/25 micrograms (see Dosage & Administration).
Following repeat dosing of fluticasone furoate/vilanterol for 7 days, there was no significant increase in systemic exposure to vilanterol (Cmax and AUC) in subjects with mild, moderate, or severe hepatic impairment (Child-Pugh A, B or C).
There were no clinically relevant effects of the fluticasone furoate/vilanterol combination on beta-adrenergic systemic effects (heart rate or serum potassium) in subjects with mild or moderate hepatic impairment (vilanterol, 25 micrograms) or with severe hepatic impairment (vilanterol, 12.5 micrograms) compared with healthy subjects.
Other special populations: In subjects with asthma, estimates of fluticasone furoate AUC(0-24) for East Asian, Japanese and South East Asian subjects (12-13% of subjects) were on average 33% to 53% higher compared with other racial groups. However, there was no evidence for the higher systemic exposure in this population to be associated with greater effect on 24-hour urinary cortisol excretion. On average, vilanterol Cmax is predicted to be 220 to 287% higher and AUC(0-24) comparable for those subjects from an Asian heritage compared with subjects from other racial groups. However, there was no evidence that this higher vilanterol Cmax resulted in clinically significant effects on heart rate.
100/2.5 mcg: In subjects with COPD estimates of fluticasone furoate AUC(0-24) for East Asian, Japanese and South East Asian subjects (13-14% subjects) were on average 23% to 30% higher compared with Caucasian subjects. However, there was no evidence for the higher systemic exposure in this population to be associated with greater effect on 24-hour urinary cortisol excretion. There was no effect of race on pharmacokinetic parameter estimates of vilanterol in subjects with COPD.
Gender, weight and BMI: No dosage adjustment is necessary based on gender, weight or BMI.
100/2.5 mcg: There was no evidence for gender, weight or BMI (body mass index) to influence the pharmacokinetics of fluticasone furoate based on a population pharmacokinetic analysis of phase III data in 1213 subjects with asthma (712 females) and 1225 subjects with COPD (392 females).
There was no evidence for gender, weight or BMI to influence the pharmacokinetics of vilanterol based on a population pharmacokinetic analysis in 856 subjects with asthma (500 females) and 1091 subjects with COPD (340 females).
200/2.5 mcg: There was no evidence for gender, weight or BMI (body mass index) to influence the pharmacokinetics of fluticasone furoate based on a population pharmacokinetic analysis of phase III data in 1213 subjects with asthma (712 females).
There was no evidence for gender, weight or BMI to influence the pharmacokinetics of vilanterol based on a population pharmacokinetic analysis in 856 subjects with asthma (500 females).
Toxicology: Preclinical safety data: Pharmacological and toxicological effects seen with fluticasone furoate or vilanterol in nonclinical studies were those typically associated with either glucocorticoids or beta2-agonists. Administration of fluticasone furoate combined with vilanterol did not result in any significant new toxicity.
Genotoxicity and carcinogenicity: Fluticasone furoate: Fluticasone furoate was not genotoxic in a standard battery of studies and was not carcinogenic in lifetime inhalation studies in rats or mice at exposures similar to those at the maximum recommended human dose, based on AUC.
Vilanterol trifenatate: In genetic toxicity studies, vilanterol (as alpha-phenylcinnamate) and triphenylacetic acid were not genotoxic indicating that vilanterol (as trifenatate) does not represent a genotoxic hazard to humans.
Consistent with findings for other beta2-agonists, in lifetime inhalation studies vilanterol trifenatate caused proliferative effects in the female rat and mouse reproductive tract and rat pituitary gland. There was no increase in tumour incidence in rats or mice at exposures 1.2- or 30-fold, respectively, those at the maximum recommended human dose, based on AUC.
Reproductive toxicity: Fluticasone furoate: Effects seen following inhalation administration of fluticasone furoate in combination with vilanterol in rats were similar to those seen with fluticasone furoate alone.
Fluticasone furoate was not teratogenic in rats or rabbits, but delayed development in rats and caused abortion in rabbits at maternally toxic doses. There were no effects on development in rats at exposures approximately 3-times greater than those at the maximum recommended human dose, based on AUC.
Vilanterol trifenatate: Vilanterol trifenatate was not teratogenic in rats. In inhalation studies in rabbits, vilanterol trifenatate caused effects similar to those seen with other beta2-agonists (cleft palate, open eyelids, sternebral fusion and limb flexure/malrotation). When given subcutaneously there were no effects at exposures 84-times greater than those at the maximum recommended human dose, based on AUC.
Neither fluticasone furoate nor vilanterol trifenatate had any adverse effects on fertility or pre- and post-natal development in rats.